细胞和基因疗法制造:临床转化中的挑战
来源:生物制品圈 | 发布时间:2024-10-21
摘要:细胞和基因疗法的安全性和有效性已在众多的临床前和临床试验中得到证明。嵌合抗原受体T细胞(CAR-T)疗法,利用了细胞和基因疗法的技术,也显示出了治疗各种癌症的巨大潜力。相关领域的进展也凸显了在制造细胞和基因疗法产品时面临的挑战。必须解决潜在的问题和障碍,以促进个体疗法的临床转化。关于代表性的基于细胞的、基于基因的以及基于细胞的基因疗法的文献回顾,涉及它们的通用制造过程、制造过程中面临的挑战以及质量控制(QC)规范是有限的。我们回顾了细胞和基因疗法的通用制造过程,包括涉及间充质干细胞、病毒载体和CAR-T细胞的过程。与制造过程相关的复杂性以及随后的QC/验证过程可能带来的挑战,可能会阻碍产品的临床进展。本文讨论了这些潜在的挑战。此外,我们讨论了制造模型的使用及其对细胞和基因疗法的影响。

引言
近年来,随着美国食品药品监督管理局(U.S. FDA)批准的细胞和基因治疗(CGT)产品数量逐年增加,对CGT的需求急剧上升。这种日益增长的需求强调了制造商迫切需要改进和推进他们的生产流程,以跟上快速发展的CGT领域。整个CGT制造过程必须在一个符合良好生产规范(GMP)规定的设施中进行,并且已经获得了全面授权,用于生产CGT或在欧洲被称为先进治疗药物产品,供人类使用。已经进行了许多临床试验,以评估CGT在包括癌症在内的广泛临床适应症中的安全性和有效性。CGT包括将细胞、基因或基因修饰细胞注入患者体内,目的是预防或治疗疾病。细胞治疗涉及注入细胞以替代或帮助修复受损细胞,而基因治疗涉及将基因注入细胞以改变受体细胞的遗传构成。基于细胞的基因疗法,如嵌合抗原受体T(CAR-T)疗法,代表了两种方法的结合。
根据彭博情报和国际细胞与基因治疗协会商业化委员会最近对CGT投资者进行的一项调查,临床显著数据是CGT领域投资者首要考虑的因素。在制造过程中保持细胞和/或基因治疗产品的效力是可能影响整体临床结果的关键因素。在制造治疗产品时面临的挑战必须得到解决,以确保其在临床上的有效转化。在这篇综述中,我们提供了代表性CGT的概述,即用于细胞治疗的间充质干细胞(MSCs)、用于基因治疗的慢病毒载体(LVs)和腺相关病毒载体(AAVs),以及用于基于细胞的基因治疗的CAR-T细胞。我们还总结了CGT的通用制造流程,并强调了在制造过程中可能遇到的挑战,这些挑战可能会阻碍相应CGT的临床使用。
间充质干细胞(MSC)基础的细胞疗法
在涉及MSC基础细胞疗法的临床试验中,已经研究了包括T细胞、干细胞、树突细胞和自然杀伤(NK)细胞在内的各种细胞类型。癌症是白细胞(T细胞、树突细胞和NK细胞)疗法的主要适应症。在白细胞之后,使用最广泛的第二种细胞疗法是干细胞基础疗法,涉及造血干细胞(HSCs)或MSCs。尽管已经为MSC疗法执行了众多自体干细胞移植,但同种异体细胞主要用于此目的。MSCs容易获得,可以从不同的来源获取,包括骨髓、脂肪组织和脐带组织。MSCs的作用机制归因于旁分泌活动,而不是直接替代受损细胞。旁分泌活动指的是MSCs向微环境中分泌旁分泌因子的能力。
MSC制造过程的上游(图1)始于组织采集,然后从组织来源分离MSCs。根据组织来源的不同,采用不同的技术从组织中分离MSCs。细胞在通常含有补充物(如胎牛血清FBS)的传统培养基中培养。然后,MSCs在不同的规模上扩增,范围从多层培养瓶到生物反应器。培养持续时间和传代次数因制造商和机构而异。通过QC分析确认细胞的身份、效力和安全性。表征MSCs的最低标准包括(但不限于)它们的身份、无菌性、活性、纯度、效力和效果。为了准备和分发注射用细胞,它们在扩增后被收获,并经过以下步骤:配方、灌装和完成、冷冻、储存和运输。
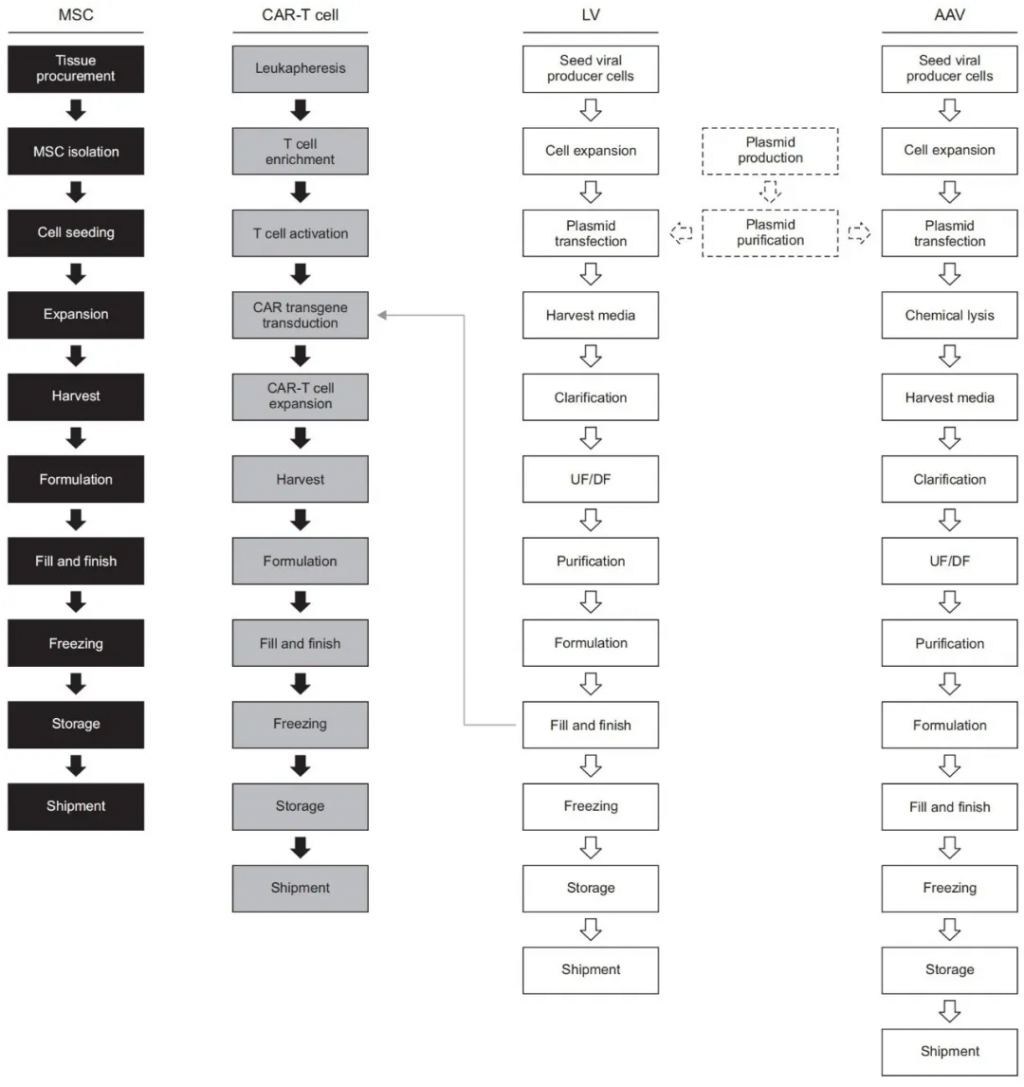
图1. 细胞和基因治疗制造流程。黑色框表示基于间充质干细胞的细胞治疗流程图。灰色框表示基于CAR-T细胞的基因治疗流程图。基于慢病毒载体(LV)和腺相关病毒(AAV)的基因治疗流程用白色实线框表示。质粒的生产和纯化用白色虚线框表示。
MSC制造中面临的常见挑战见表1。为了达到临床剂量,必须广泛扩增MSCs。然而,在扩增过程中,可能会因供体、组织来源或起始材料的条件/无菌性、分离技术和培养方法的不同而出现变异性。这些因素影响MSCs的异质性和效力。MSCs的连续传代和长期培养也可能影响它们的效力,特别是它们的增殖能力。延长MSC培养时间意味着使用更多的培养基和补充物,如FBS,从而提高整体制造成本。此外,增加对FBS的暴露可能会放大现有的安全问题,包括在受体中引起的不良免疫反应和FBS的高批次间变异性。为了确保MSC基础细胞疗法在临床环境中的进展,至关重要的是要采用能够实现快速、大规模MSC扩增的方法,平衡细胞的治疗效力与可接受的制造费用。
表1. CGT产品制造过程中可能影响其临床使用的挑战
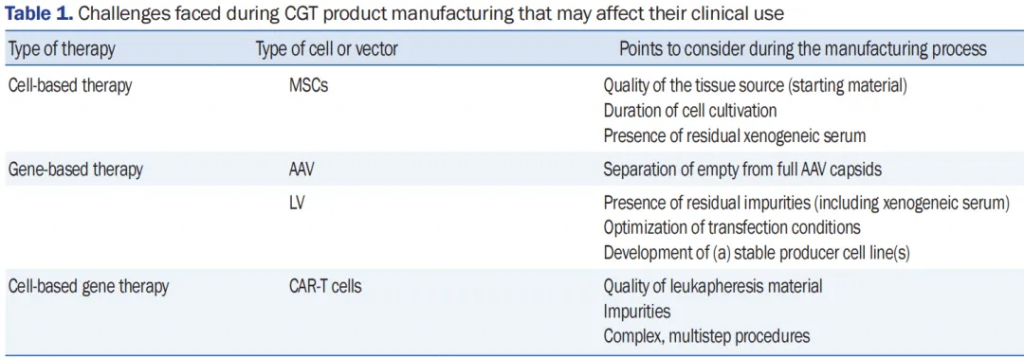
缩写词:CGT,细胞和基因治疗;MSCs,间充质干细胞;AAV,腺相关病毒;LV,慢病毒载体;CAR-T,嵌合抗原受体T细胞。
基于AAV和LV的基因疗法
AAVs和LVs是用于基因疗法的重要病毒载体。目前,FDA批准的、商业上可用的基于AAV的疗法针对视网膜营养不良和脊髓萎缩等疾病,而基于LV的基因疗法针对B细胞淋巴瘤、β地中海贫血和脑肾上腺白质营养不良等疾病。这两种载体都包装基因,并使感兴趣的基因直接给患者使用。两种载体之间的一个主要区别是AAVs包装单链DNA,而LVs包装RNA基因组。AAVs的优势包括低免疫原性、高传递和效率以及长期表达。LVs的优势包括长期转基因表达和转导非分裂细胞的能力。LVs永久性地整合到宿主基因组中。尽管总体频率较低,但AAVs也可能发生基因组整合。
基于质粒的方法通常用于制造AAVs。上游AAV制造过程(图1)从大规模质粒生产和纯化开始。必须准备以下质粒以进行转染:(1) 携带感兴趣治疗基因的顺式作用质粒,(2) 包含AAV复制和衣壳形成所需蛋白编码基因的反式作用质粒,以及(3) 使AAV在宿主细胞中复制的反式作用质粒。所有三种质粒都被转染到AAV生产细胞系中,例如人胚肾293(HEK293)细胞,以生产病毒载体。在大规模扩增(在一次性生物反应器中生长)之前,从细胞库中解冻细胞储备并培养。在进行化学裂解后,这涉及破坏细胞以将AAVs释放到培养基中,进行核酸酶处理以通过选择性降解DNA杂质来纯化载体。随后,收获的病毒上清液经过包括离心、层析和超滤(UF)和透析(DF)的纯化过程,以去除细胞碎片和杂质。杂质包括质粒DNA、蛋白质和来自AAV生产细胞的DNA以及空衣壳。
必须进行QC检测以评估病毒载体的身份、效力和安全性。QC测试的例子包括检测病毒污染、残留DNA和残留蛋白质,以及测量载体基因组浓度。最终制造步骤包括配方后进行灌装和完成、冷冻、储存和运输。实施处理、过滤和/或离心方法以从病毒上清液中去除杂质。LVs的最终配方在最终灌装和完成步骤中实现。对于AAVs,已经为LVs建立了QC要求,包括测试如评估它们的物理和功能滴度以及检测和去除杂质。
上游LV制造过程(图1)从培养和大规模扩增(即,在生物反应器中)开始,用于生产LV的细胞系,例如HEK293T,这是表达SV40 T抗原的HEK293细胞系的衍生物。SV40 T抗原表达可防止先天免疫反应的激活。HEK293T细胞也可用于以高滴度生产LVs。然而,SV40 T抗原的存在引起了关于临床制造的安全性问题。因此,正在开发不表达SV40 T抗原的LV生产细胞用于临床目的。此外,由于对使用异种血清(FBS)和在纯化过程中去除残留血清的担忧,已经实施了成熟的方法,在无血清培养基中培养LVs,其产量与含血清补充培养基相当。使用转染剂可在HEK293T细胞中瞬时表达编码感兴趣转基因和泡状口炎病毒G蛋白、包膜、gag和rev基因的质粒。通常,细胞与三到四个质粒共转染。与AAVs不同,大多数LV载体从细胞中释放出来,培养上清液用于下游过程。
尽管普遍使用,瞬时表达可能不是大规模制造AAVs和LVs的理想选择,因为存在可扩展性问题。为了扩大瞬时表达系统,需要大量的转染试剂和遗传物质(DNA),这导致生产成本增加,并且可能,批次间变异性。一个替代选择是建立和使用稳定的生产细胞系。稳定生产细胞系的生产需要使用辅助病毒,如腺病毒。但它不需要质粒DNA,因为病毒载体生产所需的基因已经整合到细胞系中。使用稳定细胞系可以产生较少的空衣壳,并且比瞬时表达更好的载体质量。使用稳定生产细胞系还可以降低制造成本,最大化AAVs和LVs的滴度。因此,使用稳定生产细胞系可以帮助克服目前基因治疗面临的挑战,例如产品产量低。
表1中展示了AAV和LV制造中面临的挑战。纯化是AAV和LV都面临的主要障碍。AAV和LV的纯度取决于载体生物制造过程,残留杂质可能影响最终产品产量。开发一种有效、可扩展的纯化方法对于降低符合GMP的生产总成本和增加载体产量至关重要。空满分离(即,将空衣壳和部分衣壳与满AAV衣壳分离)是另一个挑战,可能影响基于AAV疗法的临床使用。空AAV衣壳的污染可能在宿主中产生免疫反应,例如增强T细胞增殖。转染条件必须优化以最大化AAV和LV的生产。尽管使用稳定的生产细胞系可能服务于临床有益的目的,但在制造过程中必须考虑几点以促进临床转化,例如与辅助病毒的污染或由AAV生产所需的载体组分引起的细胞毒性。纯化过程是LV制造中的另一个关键步骤,因为杂质(如残留DNA、HEK293T细胞蛋白、培养基成分和转染试剂)可能产生不必要的炎症反应。LV固有的不稳定性质是生产过程中的主要挑战,其中环境因素可以影响并废除LV的功能。
CAR-T细胞基础的基因疗法
CAR-T细胞的生产(图1)需要细胞和基因制造。主要用于癌症免疫疗法,作为B细胞淋巴瘤和白血病的治疗选择,CAR-T细胞疗法涉及体外遗传修饰T细胞,这使得在将CAR-T细胞输注到患者体内后能够靶向并消除癌细胞。T细胞可以遗传工程化以识别B细胞成熟抗原(BCMA)或B细胞表面抗原,如CD19和CD20。为了避免正常细胞中的靶向毒性,CAR-T细胞靶点应该是高度选择性的。
CAR-T细胞制造从白细胞分离术开始,即从患者(自体)或健康供体(同种异体)的血液中收集白细胞。然后处理白细胞分离材料以富集和分离T细胞。必须准备病毒载体,通常使用LVs或逆转录病毒载体,与T细胞一起,将CAR转基因转导到后者。就临床应用而言,LVs是有吸引力的候选者,因为它们比其他载体更有效地转导缓慢增殖或不增殖的细胞。抗原呈递细胞(如树突细胞)用于体内激活T细胞,促进它们的刺激增殖和分化。就临床制造的目的而言,激活T细胞的最常见方法涉及使用单克隆抗体(如抗CD3抗体)和白细胞介素(如白细胞介素-2)。激活的T细胞通过LVs与CAR转基因转导并在培养容器中扩增。必须考虑在培养基中使用血清。以前的报告表明,血清可以影响T细胞的功能,并且使用无血清培养基增强了T细胞和CAR-T细胞在体外和体内的杀伤潜力。在配方和灌装后,CAR-T细胞制造的最后步骤包括冷冻、储存和运输。
起始白细胞分离材料的质量必须经过严格审查,以提高CAR-T细胞治疗在临床阶段的效力和效力。对于CAR-T细胞治疗,必须考虑分离的T细胞和LVs的纯度和效力。选择和分离T细胞的技术必须优化,以确保使用纯净的T细胞群体作为起始材料。起始材料中残留的污染单核细胞会干扰T细胞的激活和转导,可能导致T细胞质量差和CAR-T细胞制造失败。除了确认T细胞的纯度外,CAR-T细胞的QC要求包括检查细胞表面CAR表达和无菌性测试(即效力测试),这些测试必须快速进行,因为CAR-T细胞通常在生产后不久就会输注。
在制造过程中必须考虑T细胞收集和CAR-T细胞输注之间的时间(即静脉至静脉时间)。静脉至静脉时间的优化对于需要紧急治疗的患者至关重要。据报道,包括制造和质量评估所需的时间在内的静脉至静脉时间是三到五周。始终存在患者病情恶化和疾病进展不利的担忧。CAR-T细胞制造正转向使用封闭的、自动化的、一次性的、符合GMP的系统,该系统可以在两周内产出CAR-T细胞产品。这一进步将为生产CAR-T细胞提供标准化方法,并消除手动处理或操纵对制造的CAR-T细胞产品整体质量的影响。
质量控制测试
如前所述,CGTs的整体制造过程是艰巨和复杂的。执行适当的过程中QC和验证测试对于将各自的治疗产品转化为临床使用至关重要。不同CGTs通常检查的几个属性,如身份、纯度、效力和安全性(无菌性和存在外来因子、内毒素或支原体),在表2中列出了用于评估质量属性的各种测试和测量方法。考虑到CAR-T细胞治疗的时间重要性,制造中应实施高效和快速的QC测试。目前现有的核酸扩增测试可以比传统方法更快地检测支原体感染。证明病毒载体没有被复制能力病毒(RCVs)污染对于相关治疗至关重要,因为RCVs可能影响病毒载体的行为。
表2. 质量控制(QC)规范的测量
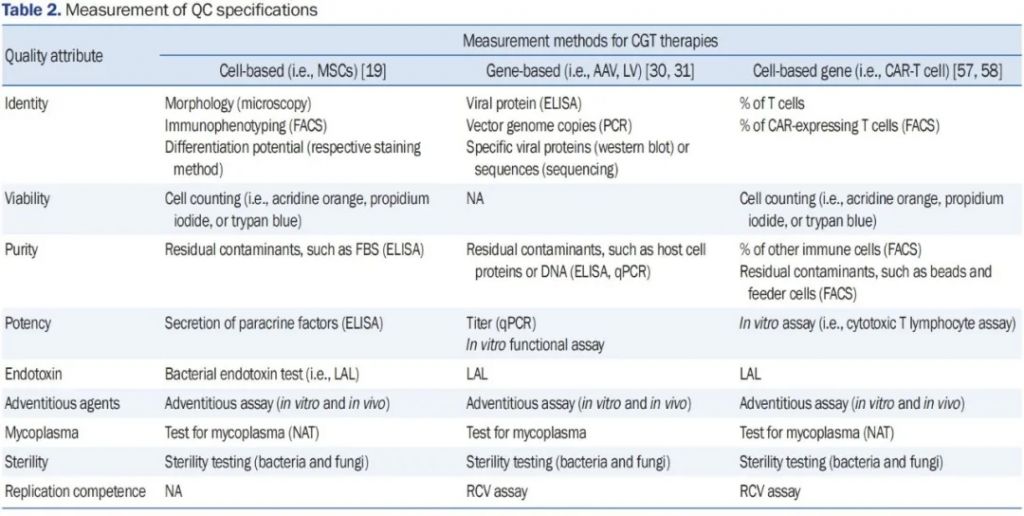
缩写词:CGT,细胞和基因治疗;MSCs,间充质干细胞;AAV,腺相关病毒;LV,慢病毒载体;CAR-T,嵌合抗原受体T细胞;FACS,荧光激活细胞分选;CAR,嵌合抗原受体;FBS,胎牛血清;qPCR,定量聚合酶链反应;LAL,鲎试剂;NAT,核酸扩增检测;NA,不适用;RCV,复制能力病毒。
每个制造商都有必须满足的规格才能发布成品。表3列出了四种代表性的FDA批准的CAR-T细胞产品的公开披露批次放行规格,即Kymriah、Yescarta、Tecartus和Carvykti。Kymriah、Yescarta和Tecartus针对CD19,而Carvykti针对BCMA。尽管Kymriah针对CD19,Carvykti针对BCMA,但两种产品都验证了CAR的存在以确认其身份。Kymriah的效力测试侧重于干扰素-γ释放,而Carvykti测试侧重于活T细胞中的CAR表达,以及一个未披露的组成部分。尽管Kymriah、Yescarta和Tecartus都针对CD19,但在最终产品发布前使用不同的方法评估其质量和安全性。在纯度评估方面存在主要区别。Tecartus的纯度规格不可用,无法直接与其他针对CD19的CAR-T细胞治疗进行比较。然而,用于Kymriah和Yescarta的评估方法差别很大。测试Yescarta的纯度时,评估了庆大霉素、内毒素和未披露的试剂,而Kymriah则对活T细胞或CD19阳性B细胞的百分比进行了量化。
表3. 四种FDA批准的CAR-T疗法的批次放行规格
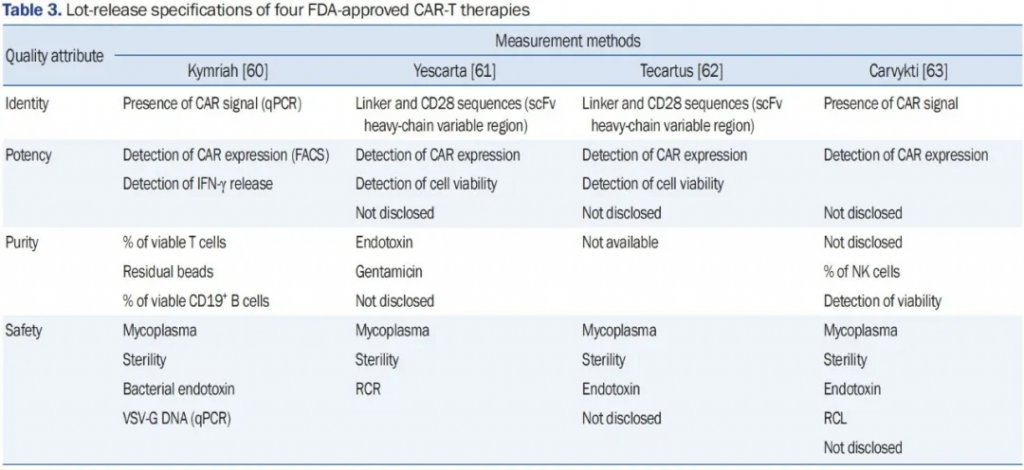
缩写词:FDA,美国食品药品监督管理局;CAR-T,嵌合抗原受体T细胞;CAR,嵌合抗原受体;qPCR,定量聚合酶链反应;scFv,单链可变片段;FACS,荧光激活细胞分选;NK,自然杀伤细胞;VSV-G,泡状口炎病毒糖蛋白G;RCR,复制能力逆转录病毒;RCL,复制能力慢病毒。
不同FDA批准的CAR-T细胞治疗使用的变化方法评估质量和安全性强调了标准化测试方法的必要性。美国FDA关于开发CAR-T细胞治疗的行业指南草案为关键质量属性的测试开发和评估方法提供了潜在的有价值的见解,包括身份和效力。关于身份,建议使用流式细胞术或PCR检测转基因。该文件建议使用细胞表面标记物观察最终产品的细胞组成,并强调测试两种载体和CAR-T细胞的效力。它还强烈建议使用涉及各种分析的矩阵方法,如细胞杀伤分析、细胞因子分泌分析和转导效率测量来确认效力。
未来展望
在CGT领域,确定集中式还是分散式制造是更合适的制造模型是一个有争议的话题。集中式制造涉及在集中的GMP设施中生产产品,并将产品分发到护理点。采用分散式制造时,如果GMP设施靠近护理点,就可以实现本地生产,这大大减少了产品交付所需的时间。将分散式模型应用于基于细胞的治疗,如CAR-T细胞治疗,将是有利的,考虑到快速向癌症患者交付产品至关重要。然而,促进多中心制造将需要标准化制造协议,以最小化产品变异。
无论实施哪种制造模型,都必须优先考虑患者的可及性。医院的GMP设施内的内部制造代表了集中制造方法,并确保了患者的可及性。医院内的内部制造可以帮助克服制造CGTs用于临床使用时面临的许多挑战。通过从手术室快速交付到GMP设施,可以确保组织来源(起始材料)的质量,这将减少制造和临床应用之间的时间延迟,并实现在单一地点并行生产和患者监测。
医院内内部制造的这些特点将惠及涉及干细胞和CAR-T细胞治疗的所有同种异体和自体治疗。如果医院没有GMP设施,CAR-T细胞生产必须外包,涉及当地细胞收集、冷冻保存和运输到制造现场。外包制造过程(包括QC),然后是最终产品运回护理点,增加了静脉至静脉的时间。现实世界分析表明,目前可用的代表性CAR-T细胞产品的静脉至静脉时间超过28天。医院内的GMP内部制造可能成为减少静脉至静脉时间的解决方案。
高成本和GMP监管合规性只是医院在建立GMP设施时面临的多重挑战之一。医院必须能够支付建设和维护制造设施的成本,设施必须遵守GMP法律法规。如果医院无法克服与建立内部GMP设施相关的障碍,另一种选择是它们开始一个衍生公司。这些衍生公司可以与当地医院保持强大的网络,并专门满足其制造需求。理想情况下,衍生公司将提供一站式购物服务,细胞、基因和基于细胞的基因治疗的生产和制造发生在靠近护理点和最重要的患者的单一地点。
结论
CGT是一个相关的话题,因为它代表了治疗广泛疾病的一种有前途的策略。对CGTs的日益关注突出了制造过程的复杂性,这可能阻碍了制造产品在临床应用中的使用。要从展示潜力到展示强大的临床效力,解决和克服与CGTs相关的制造挑战是必要的。