人工智能如何助力小分子药物开发
来源:药渡 | 发布时间:2023-12-27
摘要:尝试应用人工智能促进药物发现的初创公司,如同雨后春笋一般不断涌现,目前活跃在药物发现领域的公司至少超过350家。其中包括早期阶段的公司、初创公司以及较成熟的公司和IPO阶段的公司。最近这一领域出现了一波突破,人工智能系统帮助快速发现和开发一流的小分子,并且这些小分子都进入了临床研究,以Insilico Medicine,Exscientia,BenevolentAI,Recursion Pharmaceuticals,Deep Genomics等公司作为代表。人工智能在小分子药物发现中具有一些独特的优势,这些优势使得它成为这一领域的有力工具。
人工智能已经在药物设计与开发方面,得到了广泛的重视。尝试应用人工智能促进药物发现的初创公司,如同雨后春笋一般不断涌现,目前活跃在药物发现领域的公司至少超过350家。其中包括早期阶段的公司、初创公司以及较成熟的公司和IPO阶段的公司。最近这一领域出现了一波突破,人工智能系统帮助快速发现和开发一流的小分子,并且这些小分子都进入了临床研究,以Insilico Medicine,Exscientia,BenevolentAI,Recursion Pharmaceuticals,Deep Genomics等公司作为代表。
值得注意的是,大多数人工智能驱动的初创公司都专注于小分子药物的发现,而不是生物制剂,这一点其实并不令人感到意外。从历史上看,即使是非人工智能计算方法(化学信息学)也主要用于小分子治疗,因为它们的分子结构和相互作用模式要简单得多。
人工智能在小分子药物发现中具有一些独特的优势,这些优势使得它成为这一领域的有力工具:
01化学信息可用性
小分子药物的发现通常涉及大量的化学信息,包括分子结构、药物活性和毒性等。这些信息以数字形式存在,易于被计算机处理和分析。相比之下,生物制剂(如蛋白质药物)的发现和设计可能涉及更为复杂的生物学信息,难以用数字表示,因此处理起来更为复杂。
02大规模虚拟筛选
小分子药物的发现可以通过大规模的虚拟筛选来加速,这正是人工智能的一个强大优势。AI可以处理庞大的化学数据库,预测分子的药效、毒性和其他关键特性,从而更高效地筛选出潜在的候选化合物。生物制剂的发现通常涉及更少的分子,且虚拟筛选的规模相对较小。
03化学空间探索
人工智能能够有效地探索和理解化学空间,推动新化合物的设计和创新。在小分子药物的领域,通过学习大量的已知药物和相关化合物的数据,AI可以帮助挖掘新的化学结构,从而促进新药物的开发。相比之下,生物制剂的设计通常更依赖于生物学信息。
04高通透性和生物利用度的优化
小分子药物需要具有良好的口服吸收特性,这涉及到化合物的物理化学性质。AI在优化小分子药物的通透性和生物利用度方面有一定优势,因为这些特性可以通过大量的实验数据进行建模和预测。
这些优势使得人工智能在小分子药物发现中能够更迅速、精准地筛选和设计化合物,加速新药物的发现和开发过程。
与之相对应的是,生物制剂在利用人工智能进行药物发现的过程中,相对于小分子药物,可能具有以下不利因素:
01复杂性和多样性
生物制剂通常是大型复杂的蛋白质、抗体或其他大分子,其结构和功能更加复杂多样。处理这种复杂性数据需要更高级的模型和算法,而当前的AI技术在处理大规模多样性的生物制剂数据时,可能面临一些挑战。
02数据可用性
生物制剂的研发,涉及到数据库在不同生物系统中的复杂相互作用。尽管生物制剂的数据逐渐增多,但相较于小分子药物,可用于训练AI模型的大规模生物制剂数据仍然有限,限制了模型的学习和预测能力。
03定制性和个性化
生物制剂通常具有较高的定制性和个性化,因为它们是根据具体疾病或患者需求设计的。这增加了预测和设计的难度,因为每种生物制剂可能具有不同的结构、功能和相互作用模式。
04生物学多样性
生物制剂往往通过与人体的生物系统相互作用来实现治疗效果,而人体的生物学多样性和复杂性,使得生物制剂的研发更为具有挑战性。AI模型需要更好地考虑这些多样性,以实现更准确地预测生物制剂在不同个体中的效果。
人工智能在药物分子发现的过程中,得到了非常广泛的应用,仅次于疾病建模和靶标发现。人工智能驱动的药物设计,主要分为三大类:从头药物设计、现有数据库的虚拟筛选、药物再利用。
图1. AI药物分子发现的三大类别
01从头药物设计
从头药物设计(de novo drug design)主要是通过深度学习模型实现的,例如生成对抗神经网络(GAN,generative adversarial neural networks,是一类深度学习模型,由生成器和判别器组成。GAN的训练过程是一个博弈,通过对抗的方式不断提升生成器和判别器的性能)。生成式人工智能平台的一些例子,包括Insilico Medicine的Chemistry42软件、Iktos的Makya和Ro5的De Novo Platform。还包括 Recursion Pharmaceuticals、Deep Cure、Standigm等。
简单地说,de novo drug design是一种通过计算机辅助的方法,来设计全新的药物分子。人工智能在de novo药物设计中扮演着关键的角色,可以通过以下步骤进行:
- 数据收集:首先,系统需要大量的生物化学和药理学数据,包括已知药物的结构、活性、毒性等信息。这些数据用于训练机器学习模型。
- 特征提取:在训练模型之前,需要从收集的数据中提取特征,这些特征可能包括分子的结构、电荷分布、溶解度等。这一步骤的目的是将化学信息转化为计算机能够理解的数字形式。
- 机器学习模型训练:采用各种机器学习算法,例如深度学习或基于规则的方法,训练模型以理解药物分子的结构和活性之间的关系。这使得模型能够从已知数据中学到一般的规律。
- 生成新分子:一旦模型训练完成,它就可以用于生成新的、未见过的分子结构。这可以通过从随机的分子结构开始,然后通过模型的生成能力不断优化,直到达到满足特定目标的药物分子。
- 评估和筛选:生成的分子结构需要经过评估,以确保其具有潜在的药用价值。这可能涉及对生物活性、毒性、生物可用性等方面的预测。
- 优化和合成规划:生成的分子通常需要进行进一步的优化,以提高其在实验中的合成可行性和生物活性。AI还可以提供合成规划,帮助确定实验室中如何制备这些新分子。
整个过程是一个迭代的循环,通过不断优化模型并尝试新的分子设计,最终目标是找到具有良好生物活性和临床潜力的新药物分子。这种方法能够加速药物发现的过程,尤其是在探索大量的潜在分子结构时,AI的高效性体现得尤为明显。
02虚拟筛选
应用人工智能进行药物分子发现的第二个途径是超大规模虚拟筛选,筛选数十亿个分子以找到成功的目标。2022年8月,赛诺菲与Atomwise合作进行了一项价值可能高达12亿美元的药物设计交易。赛诺菲预付2000万美元,重点是利用Atomwise的 AtomNet平台,来研究赛诺菲选择的多达5个药物靶点的小分子。公告称,基于卷积神经网络的AtomNet擅长基于结构的药物设计,能够“通过人工智能快速搜索 Atomwise超过3万亿种可合成化合物”。
虚拟筛选是一种利用计算机模型和算法对潜在药物分子进行预测和评估的方法,以便从大量的化合物库中,筛选出具有潜在生物活性的候选分子。这种筛选过程是通过在计算机中进行模拟和预测,而不是在实验室中进行物理实验来完成的,因此称之为”虚拟”筛选。
虚拟筛选的主要目标是在药物发现的早期阶段,从数百万到数千万个潜在的药物候选分子中,鉴定出可能对特定疾病目标具有生物活性的分子。这有助于加速药物研发过程,减少实验室实验的时间和成本。在虚拟筛选中,人工智能的应用主要通过构建预测模型来实现:
- 数据预处理:进行数据清洗、去噪声和标准化。这确保了模型训练所使用的数据是准确且一致的,提高了模型的性能。
- 分子表征:将分子结构转换为计算机可处理的特征表示形式。这可以通过分子描述符(molecular descriptors)、分子指纹(molecular fingerprints)、图神经网络(graph neural networks)等方法来实现。合适的分子表征对于模型性能至关重要。
- 模型选择:选择适当的机器学习或深度学习模型。常用的模型包括支持向量机、随机森林、深度神经网络等。选择的模型应根据任务的性质和数据的特点来进行。
- 模型训练:使用已知生物活性的化合物数据集对选定的模型进行训练。这样,模型能够学习药物分子的结构与生物活性之间的关系。
- 模型评估:利用验证集进行模型的评估,以检验其对未见过数据的泛化能力。评估指标可能包括准确性、灵敏度、特异度等。
- 虚拟筛选:使用训练好的模型对潜在的药物分子进行预测。这可以是从公共数据库中获取的已知分子,也可以是通过计算或合成生成的新分子。模型会为每个分子提供一个生物活性的预测值,根据这些预测值进行排序。
- 分子优化:对于在虚拟筛选中排名较高的分子,可能需要进一步的化学优化。这可以通过调整分子结构以提高生物活性、改善药代动力学性质等来实现。
- 实验验证:虚拟筛选的结果需要在实验室中进行验证。实验验证有助于确认虚拟筛选的准确性,并验证潜在药物分子的生物活性和其他关键性质。
03药物再利用
最后,许多公司正在使用药物再利用(Drug Repurposing)策略,来进行人工智能药物发现。此类公司包括Healx、BenevolentAI、BioXcel Therapeutics。他们主要使用自然语言处理 (NLP) 模型和机器学习,通过分析大量非结构化文本数据(研究文章和专利、电子健康记录以及其他类型数据),来构建和搜索可以实现再利用的药物群体。
- 数据整合与挖掘:AI可以整合和挖掘大量的生物医学数据,包括基因组学、蛋白质组学、药物相互作用等信息。这有助于发现已有药物在新的治疗领域可能具有活性的迹象。
- 网络分析:利用网络分析技术,AI可以建立药物、蛋白质和疾病之间的关联网络。通过分析这些网络,可以识别潜在的药物再利用机会,例如,发现与目标疾病相关的已有药物或化合物。
- 药物相似性和特征学习:AI可以利用药物相似性和特征学习方法,分析已有药物与新治疗目标之间的相似性。这有助于预测已有药物是否对新的治疗目标具有潜在活性。
- 机器学习预测:采用机器学习算法,AI可以根据已有药物的生物活性和药理学特性,建立预测模型,预测这些药物在新的治疗领域中的效果。这种方法有助于高效地筛选候选药物。
- 文本挖掘和知识图谱:AI技术可以通过文本挖掘和知识图谱构建,自动化地从文献、专利和临床试验数据库中,提取有关药物的信息。这有助于发现药物的新的治疗用途和关联。
- 细胞和基因组学数据分析:利用细胞和基因组学数据,AI可以识别已有药物对细胞或基因表达的影响,从而发现其可能的治疗机制,并推断其在新治疗目标上的潜在效果。
例如,美国临床阶段生物技术公司Lantern Pharma,就是这样一家专注于利用先进的基因组学、机器学习和人工智能来创新癌症药物开发流程的企业。该公司的人工智能平台——RADR®️目前包含超过250亿个数据点,并使用大数据分析和机器学习,来快速发现与药物反应相关的生物学基因组特征,然后识别相关癌症患者亚组,使其从Lantern的候选药物中受益。Lantern及其合作者还使用RADR®️来开发和定位新药以及药物再利用。
04AI在小分子药研中应用展望
根据BiopharmaTrend网站收集的数据,下图显示了319家药物发现初创公司对于人工智能的应用情况。将近一半的公司(49%,156 家初创公司)专注于小分子药物的发现,而只有20%(64家初创公司)参与发现和开发生物药物(抗体、疫苗等)。
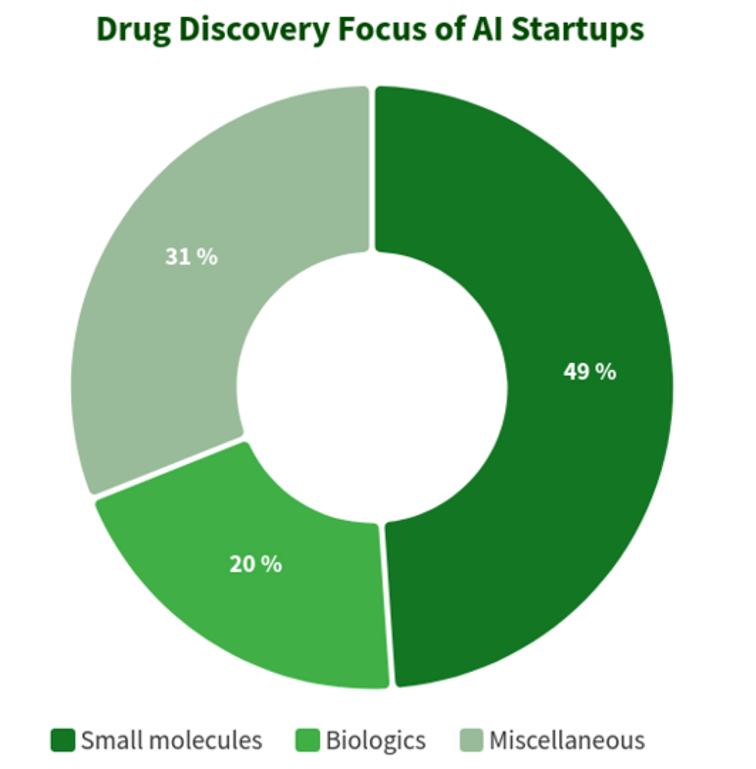
图2. 医药人工智能初创公司的业务分布图,来源:BiopharmaTrend
人工智能在小分子药物发现领域具有光明的前景,尽管目前尚没有通过人工智能实现小分子药物上市的成就,但这只是时间的问题。
人工智能最大的优势也许是极大缩短药物设计的周期。传统的药物发现过程非常耗时且昂贵,而人工智能能够加速整个过程。通过高效的虚拟筛选、分子设计和优化,人工智能可以在更短的时间内生成和评估大量的药物候选分子。根据Nature Review Drug Discovery的报告,研究者发现多个人工智能项目在不到四年的时间内完成了整个发现和临床前的过程,而通常这个研发过程需要五至六年的时间。
人工智能可以根据已有的生物学和药理学知识,设计具有目标生物活性和更好生物可用性的分子。这包括生成新的分子结构、优化药物性质等。AI还可以分析复杂的生物学网络,预测不同药物的相互作用,从而帮助发现更有效的药物组合疗法。这对于治疗复杂疾病和抗药性的问题尤为重要。通过分析大规模的生物学和临床数据,人工智能可以帮助患者实现个性化药物治疗。根据患者的遗传信息、生物标志物和疾病特征,定制药物治疗方案,提高治疗的效果。人工智能模型可以预测药物的潜在毒性和不良反应,从而帮助在早期阶段识别潜在的安全性问题,减少候选药物的流失。与结构生物学数据的结合,可以令人工智能更准确地预测小分子与蛋白质的相互作用,从而指导药物设计的方向。而在新的药物靶点识别方面,人工智能在分析大规模生物学数据的过程中,可以通过新靶点的识别,为药物发现提供更多的选择。
这些前景表明,人工智能在小分子药物发现领域的应用有望在未来取得更多的突破,为新药物的发现和开发提供更强大、高效的工具。