2025年7月FDA新药获批速览
来源:药事纵横 | 发布时间:2025-08-29
摘要:2025 年 7 月美国 FDA 共批准5 款新药,涵盖代谢疾病、皮肤病、罕见病、癌症等领域:Sephience(sepiapterin) 获批用于 1 个月及以上苯丙酮尿症(PKU)患者,基于 3 期试验实现苯丙氨酸平均减少63% ;Anzupgo(delgocitinib) 作为首个 FDA 批准的中重度慢性手部湿疹(CHE)专属疗法,属 “first-in-class” 外用泛 JAK 抑制剂;Ekterly(sebetralstat) 是首个针对 12 岁及以上人群遗传性血管性水肿(HAE)急性发作的口服按需疗法,症状缓解中位时间仅2 小时;Zegfrovy(sunvozertinib,舒沃替尼) 为唯一获批的 EGFR 外显子 20 插入突变非小细胞肺癌(NSCLC)靶向口服药,总缓解率达46% ;Lynozyfic(linvoseltamab-gcpt) 作为首个 FDA 批准的 BCMA/CD3 双特异性抗体,用于四线及以上复发或难治性多发性骨髓瘤(MM),客观缓解率70% ,但需通过风险评估和缓解策略(REMS)项目使用。
本文简单盘点了2025年7月份FDA新药审批情况,看看有哪些药界新星闪亮登场,给生命健康带来了新希望:
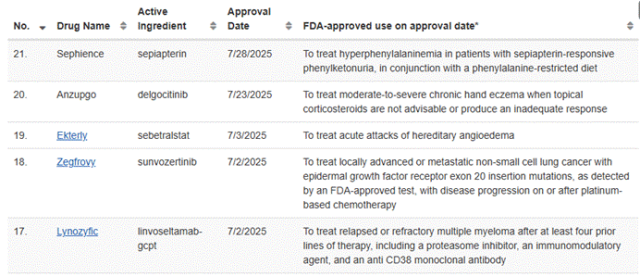
【SEPHIENCE】
公司:PTC Therapeutics
批准日期:2025年7月28日
治疗:苯丙酮尿症(PKU)
近日,PTC Therapeutics宣布,FDA已批准Sephience(sepiapterin)用于治疗儿童和成人苯丙酮尿症(PKU)患者。此次批准适应症范围广泛,适用于1个月及以上、对sepiapterin有应答的成人和儿童高苯丙氨酸血症(HPA)患者。此次FDA批准主要基于3期APHENITY临床试验中显著的疗效与安全性数据,并在APHENITY长期延伸研究中验证了治疗效果的持续性。其中APHENITY试验的结果已发表在著名医学期刊《柳叶刀》上。分析显示,接受sepiapterin治疗的患者平均苯丙氨酸减少63%。超过80%的患者血液苯丙氨酸水平降低到美国和欧盟对PKU儿童和成人患者苯丙氨酸水平的目标指标以下。
【ANZUPGO】
公司:LEO Pharma
批准日期:2025年 7月23日
治疗:中重度慢性手部湿疹(CHE)
LEO Pharma近日宣布,FDA已批准Anzupgo(delgocitinib)乳膏(20 mg/g),用于局部治疗对外用糖皮质激素应答不佳或不适合使用糖皮质激素的中重度慢性手部湿疹(CHE)成人患者。根据新闻稿,Anzupgo乳膏是专门用于治疗中重度慢性手部湿疹成年患者的首个获FDA批准的疗法。Anzupgo是一款“first-in-class”的外用泛JAK抑制剂,可抑制造成慢性炎症皮肤疾病病变的JAK-STAT信息通路的活化。
之前公布的DELTA 1和DELTA 2两项临床3期试验,评估了Anzupgo相较于安慰剂用于治疗CHE的安全性和疗效。治疗成功定义为研究者整体评估的慢性手部湿疹治疗成功(IGA-CHE TS)评分为0(皮损清除)或1(皮损几乎清除),且较基线至少提高两级。
试验数据显示,在第16周时,Anzupgo治疗组与载体对照组相比,有更大比例的患者达到IGA-CHE治疗成功的标准。DELTA 1中Anzupgo治疗组达到治疗成功标准的患者比例为20%,对照组为10%。在DELTA 2中Anzupgo治疗组达标的患者比例为29%,对照组为7%(两项试验均p≤0.0055)。
【EKTERLY】
公司:KalVista Pharmaceuticals
批准日期:2025年7月3日
治疗:遗传性血管性水肿
KalVista制药公司于7月7日宣布,FDA已批准Ekterly(sebetralstat,300mg)片剂,用于治疗成人和12岁及以上儿童患者的遗传性血管性水肿(HAE)急性发作。Sebetralstat是一种口服血浆激肽释放酶抑制剂,靶向激肽释放酶-激肽系统级联。通过抑制高分子量激肽原的裂解,该药可减少缓激肽的产生,从而缓解HAE的症状。值得注意的是,Ekterly是美国FDA批准的首个也是唯一一个用于治疗12岁及以上人群遗传性血管性水肿急性发作的口服按需疗法。
此项批准基于随机、三方交叉、3期KONFIDENT试验(ClinicalTrials.gov标识符:NCT05259917)的数据,该试验评估了Sebetralstat在12岁及以上患有1型或2型遗传性血管性水肿(HAE)的儿童和成人中的疗效和安全性。
研究参与者被随机分配接受Sebetralstat 300mg、Sebetralstat 600mg或安慰剂治疗。
研究结果显示,与安慰剂相比,Sebetralstat 600mg组患者的HAE发作症状缓解速度显著加快。在接受Sebetralstat 600mg治疗的患者中,症状开始缓解的中位时间为2小时(95% CI,1.5-2.8)。结果还显示,Sebetralstat 600mg组在首次服药后12小时内首次出现严重程度减轻的时间(使用患者总体严重程度评分[PGI-S]进行评估)显著快于安慰剂组,且具有统计学意义Sebetralstat 600mg组的严重程度减轻中位时间为9.1小时(95% CI,3.8,未达到)。
另外,Sebetralstat 600mg组的发作缓解时间(首次服药后24小时内PGI-S为“无”)显著快于安慰剂组。
【ZEGFROVY】
公司:Dizal Pharma
批准日期:2025年7月2日
治疗:表皮生长因子受体(EGFR)外显子20插入突变的局部晚期或转移性非小细胞肺癌
Dizal(迪哲医药)于7月2日宣布,FDA已加速批准Zegfrovy(sunvozertinib,舒沃替尼)用于治疗经FDA批准的检测检测出表皮生长因子受体(EGFR)外显子20插入突变的局部晚期或转移性非小细胞肺癌(NSCLC)成年患者,这些患者的病情在接受铂类化疗期间或之后出现进展。
Zegfrovy已获得FDA的优先审评和突破性疗法认定,是唯一获批的针对EGFR外显子20ins突变NSCLC的靶向口服药物。该适应症基于总体缓解率和缓解持续时间获得加速审批。该适应症的持续审批可能取决于在验证性试验中对临床获益的验证和描述。
FDA还批准赛默飞世尔科技的Oncomine Dx Express Test作为Zegfrovy的伴随诊断,以帮助检测NSCLC患者的EGFR外显子20插入突变。
Zegfrovy是由迪哲医药科学家研发的不可逆EGFR抑制剂,靶向多种EGFR突变,且对野生型EGFR具有选择性。该药物在FDA的获批基于WU-KONG1B试验(ClinicalTrials.gov注册号:NCT03974022)的数据。主要疗效人群包括85例EGFR外显子20插入突变的局部晚期或转移性非小细胞肺癌患者,且在铂类化疗期间或之后病情进展。
研究参与者每日一次口服Sunvozertinib 200mg,随餐服用,直至病情进展或出现无法耐受的毒性反应。研究结果显示,确认的总缓解率(主要终点)为46%(95% CI,35-57)。缓解持续时间为11.1个月(95% CI,8.2,无法评估)。
【LYNOZYFIC】
公司:Regeneron Pharmaceuticals
批准日期:2025年7月2日
治疗:复发或难治性(R/R)多发性骨髓瘤(MM)
再生元制药公(Regeneron Pharmaceuticals)司于7月2日宣布,FDA已加速批准Lynozyfic(linvoseltamab-gcpt)用于治疗复发或难治性(R/R)多发性骨髓瘤(MM)成年患者,这些患者既往已接受过至少四种疗法,包括蛋白酶体抑制剂、免疫调节剂和抗CD38单克隆抗体。
Linvoseltamab是一款BCMA/CD3靶向双特异性抗体,旨在将多发性骨髓瘤细胞上的BCMA与T细胞表面表达的CD3连接,以促进T细胞活化和癌细胞杀伤。Lynozyfic采用再生元的VelocImmune技术研发,是一种全人源BCMAxCD3双特异性抗体,旨在将多发性骨髓瘤细胞上的B细胞成熟抗原(BCMA)与表达CD3的T细胞连接起来,从而促进T细胞活化并杀灭癌细胞。
FDA的批准主要基于关键性1/2期LINKER-MM1试验结果,该试验评估了linvoseltamab治疗R/R MM的疗效,共有80名患者参与。由独立审查委员会评估的结果显示,患者的客观缓解率(ORR)达70%,其中45%的患者达到完全缓解(CR)或更佳的应答。患者产生首次缓解的中位时间为0.95个月(范围:0.5至6个月)。此外,中位缓解持续时间(DoR)尚未达到(95% CI:12个月至不可估计)。在中位随访13个月的应答者中,估算的9个月缓解持续率为89%(95% CI:77-95),12个月缓解持续率为72%(95% CI:54-84)。
接受推荐剂量治疗的患者中,46%出现了细胞因子释放综合征(3级,< 1%),54%出现了神经系统毒性,包括免疫效应细胞相关神经毒性(3-4级,8%)。此外,linvoseltamab-gcpt的处方信息带有关于危及生命的细胞因子释放综合征(CRS)和神经毒性(包括ICANS)的黑框警告。由于这些风险,Lynozyfic 仅通过名为 Lynozyfic 风险评估和缓解策略(REMS) 的受限项目提供。
新闻稿指出,Lynozyfic是首个获得FDA批准的BCMAxCD3双特异性抗体,可从第14周开始每两周给药一次,如果在完成至少24周的治疗后达到非常好的部分缓解(VGPR)或更佳,则每四周给药一次。该方案包括在递增剂量期间(第一次递增剂量后24小时和第二次递增剂量后24小时)住院以确保安全。
Ref:FDA官网、各药企官网新闻稿。